

Abstract
Introduction: Acute chest syndrome (ACS) is a leading cause of morbidity and death among children with sickle cell disease (SCD). Aberrant cell-cell interactions involving the endothelium are central to the pathophysiology of ACS. Endothelial integrity is partially regulated by circulating cell-derived extracellular vesicles (exosomes). We previously demonstrated that (even when at a healthy baseline) patients with SCD have increased numbers of circulating exosomes, and exosomes from SCD patients with a history of ACS cause increased disruption of endothelial integrity in vitro. The current study was designed to test the hypothesis that exosomes isolated from patients during an episode of ACS would induce much greater endothelial damage than those isolated from the same patient at their baseline.
Methods: We identified 9 patients with SCD included in our biobank who had plasma isolated at baseline and at the beginning of an admission for ACS (prior to transfusion). Samples were considered baseline if the patient was more than 4 weeks since transfusion and had no new health-related complaints. ACS was defined by the presence of an infiltrate on chest x-ray combined with fever, pain, hypoxia or cough. Control plasma samples were obtained from children without SCD or known medical problems and a BMI < 95%ile. Exosomes were isolated from plasma using established methodologies. Assessment of vesicle size was performed by nanoparticle tracking analysis. To determine the effects on endothelium, exosomes were added to cultures of human microvascular endothelial cells (HMVEC), which were subsequently studied by fluorescence microscopy (after immunolocalization of VE-cadherin, reaction with TRITC-conjugated phalloidin and treatment with DAPI). Protein components of exosomes and cells were detected and quantified by immunoblotting. VE-cadherin mRNA expression was determined by qRT-PCR. Free heme was quantified using a colorimetric assay.
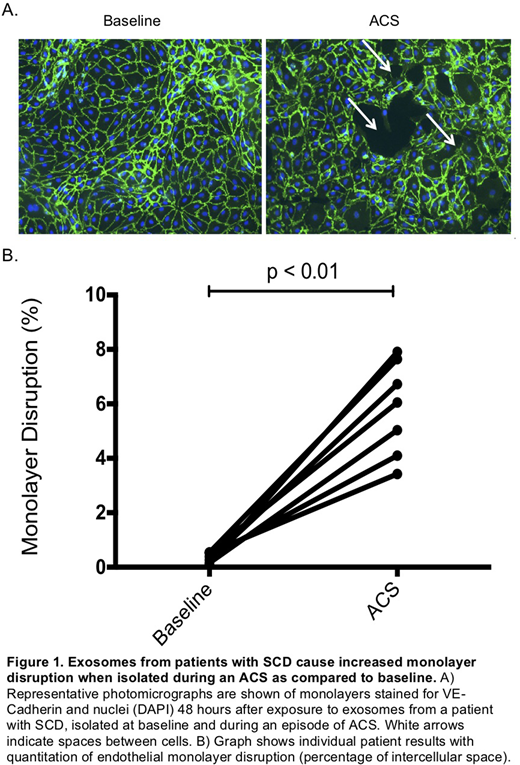
Results: Nanoparticle tracking analysis showed that all exosome preparations (regardless of patient or disease status) had a size distribution with a sharp peak of between 90 - 100 nm. Immunoblotting showed the presence of exosomal marker proteins (CD63 and flotillin) in all samples. The abundance of these proteins was increased by 50% (on average) in the samples obtained during ACS. Immunoblots showed that the exosomes also contained hemoglobin, glycophorin A, and ferritin; of these, only hemoglobin was increased in patients with SCD (2-fold greater in SCD than control) and ACS (4-fold greater in ACS than control). Heme concentration in the exosome preparations was below the assay detection limit. Microscopy showed that application of exosomes to endothelial monolayers caused no immediate damage; in addition, most samples showed no significant damage after 24 hr. However, after 48 hr, ACS-exosomes caused alterations of the localization of VE-cadherin (with opening of gaps between cells) and of actin microfilaments (as detected by phalloidin staining) consistent with cell retraction. ACS exosomes caused significantly more disruption of the monolayers than baseline-exosomes (baseline: 0.36 ± 0.07 % cell-free space vs. ACS: 5.81±0.65 % cell-free space, p < 0.01). DAPI staining showed no differences in numbers of nuclei excluding cell death or detachment. Although VE-cadherin mRNA levels were unchanged, immunoblots showed a dramatic decrease (by 62%) in VE-cadherin protein (p <0.01), in cells treated with ACS-exosomes vs. baseline-exosomes.
Conclusions: These results confirm and extend on our prior observations that circulating exosomes in SCD patients affect endothelial integrity. We confirmed that these patients have circulating vesicles consistent with designation as exosomes, based on size and the presence of marker proteins. Their composition shows that many of these exosomes must derive from red cell precursors, but they are not simply red cell-derived vesicles. ACS exosomes cause a disruption of endothelial monolayer integrity that involves rearrangement of the actin cytoskeleton and degradation of cadherin-based intercellular junctions. The time course suggests that it requires cellular signaling and perhaps new RNA/protein synthesis. We speculate that exosomes are central to the vascular pathology in all SCD patients.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal